
 教職員募集
教職員募集 所内専用
所内専用種の間で同じ遺伝子を正確に選び出すソフトウェア ORTHOSCOPE* を開発
2021年12月2日
井上 潤(東京大学大気海洋研究所 附属地球表層圏変動研究センター)
成果概要
種の間でゲノムを比較する場合、配列が類似した遺伝子が多いため、同じ機能をもった遺伝子を判定することは困難であった。東京大学大気海洋研究所の井上潤助教は、ゲノムに存在するすべての遺伝子の系統樹を推定することで、同じ機能を持った遺伝子を選び出すソフトウェア ORTHOSCOPE* (https://github.com/jun-inoue/ORTHOSCOPE_STAR![]() ) を開発した。このソフトウェアを用いた解析は、ゲノム進化の理解だけでなく、ホヤやヒトのゲノムに直接侵入したバクテリアなどに由来する遺伝子の発見、さらには環境DNA解析に有用な遺伝子マーカーの選定も可能にする。
) を開発した。このソフトウェアを用いた解析は、ゲノム進化の理解だけでなく、ホヤやヒトのゲノムに直接侵入したバクテリアなどに由来する遺伝子の発見、さらには環境DNA解析に有用な遺伝子マーカーの選定も可能にする。
発表内容
1. 研究背景
ゲノムデータ (注1) が急速に蓄積し、国際DNAデータベース NCBIでは、現在、動植物855種のゲノム配列が公開されている。まさに、生物の設計図に隠された秘密に迫る絶好の機会が到来している。このため種の間でゲノムデータを比較して機能が同じ遺伝子を判定するソフトウェアが盛んに開発されてきたが、遠縁な生物でも保存される染色体領域を明確にするなど、魅力溢れる進化解析には到達していない。既存のソフトウェアは、解析のスピード化に焦点を置き、2種を1組として配列の類似性を総当たりで比較する。このため2万以上の遺伝子からなるゲノムデータを多くの種について比較することを誇るが、異なる遺伝子を同じと判断することが極めて多い。
なぜ間違えて判定するのか。それは、遺伝子は長い生物進化の過程で重複と欠失を繰り返すため、遺伝子の間で配列が似ていても他人の空似のような状態が頻繁に見られるためだ。この状況を打開するには 50年以上に渡って培われた理論と手法からなる分子系統解析が必要なことは明確だったが、ゲノム上の個々の遺伝子について系統解析を行うには膨大な時間と作業を要することもあってか、軽視された。
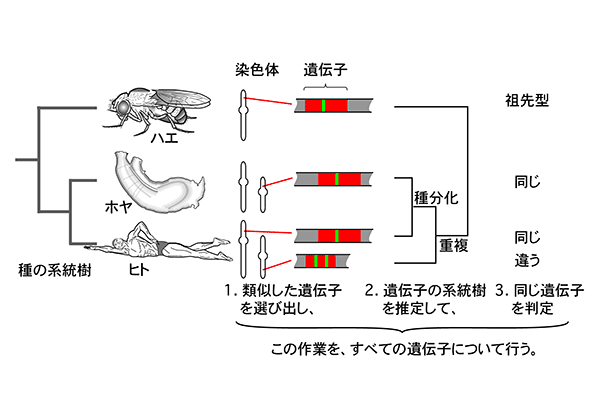
コンピューターの解析スピードが上昇した今、これらの熟成された解析をゲノム規模のデータに適用しない手はない。井上は、分子系統解析を駆使した方法 (図1) を、ORTHOSCOPE* (star) に組み込んだ。このソフトウェアは、すべての遺伝子の歴史を推定して結果を統合することで、ゲノム進化イベントを推定する。
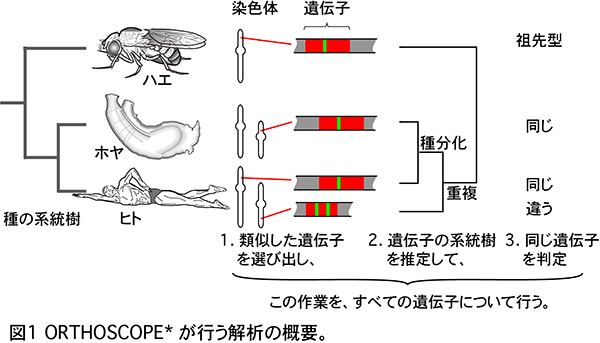
2. 研究内容
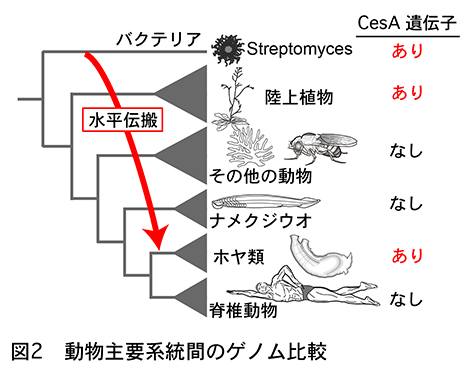
水平伝搬:ORTHOSCOPE*には、他にも画期的な特徴がある。それは、web version (ORTHOSCOPE: https://www.orthoscope.jp![]() ) から動植物550種以上のゲノムデータ (遺伝子モデル) をすぐに利用できる点である。これら ORTHOSCOPE*の能力を示すために、動物では本当にホヤ類だけがセルロース合成を担う遺伝子 (CesA遺伝子) を持つのか検証した (図2)。現生動物のCesA遺伝子は、水平伝搬 (注2) によってバクテリアからホヤ類に移動したことが知られる。ホヤ類は動物で唯一、バクテリアや植物のようにセルロースを合成する能力を獲得し体を硬い膜で覆うことが知られる。
) から動植物550種以上のゲノムデータ (遺伝子モデル) をすぐに利用できる点である。これら ORTHOSCOPE*の能力を示すために、動物では本当にホヤ類だけがセルロース合成を担う遺伝子 (CesA遺伝子) を持つのか検証した (図2)。現生動物のCesA遺伝子は、水平伝搬 (注2) によってバクテリアからホヤ類に移動したことが知られる。ホヤ類は動物で唯一、バクテリアや植物のようにセルロースを合成する能力を獲得し体を硬い膜で覆うことが知られる。
サンゴや貝類、昆虫などの動物49種を含むゲノムデータをORTHOSCOPE*で解析し、全遺伝子の歴史を推定した。するとCesA遺伝子は、ホヤ類の主要系統全てが保持しているが、他の動物にないことが再現された。このことは、バクテリアのある系統からホヤ類の祖先種に CesA遺伝子が水平伝搬したことで、ホヤ類が3000種にまで繁栄できたことを物語る。さらに新しい事実もわかった。それは、他に同様の保持パターンを示す遺伝子は存在しなかったことである。このことは、セルロース合成に利用しているCesA以外の遺伝子を、バクテリアのStreptomyces系統から得なかったことを意味する。
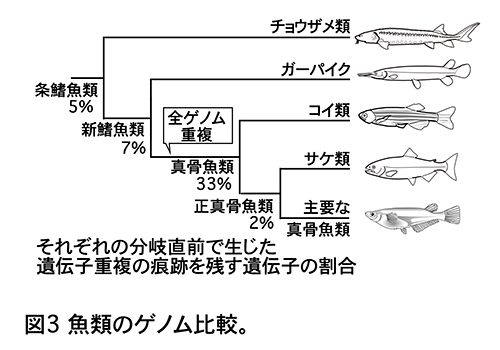
全ゲノム重複:真骨魚類は進化の初期段階に全ゲノム重複 (注3) を経験したことが知られる。この事実を ORTHOSCOPE* が検知できるか、魚類8 種のゲノムを比較して確かめた (図3)。ORTHOSCOPE*は、祖先ゲノムに存在した遺伝子の何%に、遺伝子重複の痕跡が残っていたのか算出する。すると、その割合は、確かに他の分岐よりも真骨魚類根幹の分岐の祖先ゲノム (33%) で多かった。つまり、ORTHOSCOPE*を使えば、未知の全ゲノム重複を発見する最初の手がかりが得られる。
環境DNA解析:ORTHOSCOPE*は、環境DNA解析 (注4) の指標(分子マーカー)となる遺伝子を選定できる。環境DNA解析には、これまでミトコンドリアDNAを指標としていたが、配列が酷似しているため近縁種を分けられないことが多い。このため核ゲノムにある遺伝子を指標にする必要があったが、この場合、2万もの遺伝子から適切な候補遺伝子が選ばれていなかった。そこでORTHOSCOPE*で魚類のゲノムを解析し、それぞれの種が1コピーずつ持っており、他人の空似がない1231遺伝子を候補として抽出した。さらなる検証は必要だが、これらの遺伝子のいくつかが、核遺伝子を用いた精度の高い環境DNA解析の有力な指標となるだろう。
3. 社会的意義・今後の展望
これまで行われていたゲノム比較では、遺伝子の歴史が充分に考慮されていなかったため、遺伝子にしろ染色体領域にしろ、比較する対象を間違えていることが多かった。ORTHOSCOPE*を用いれば比較する遺伝子を正しく選べるため、これまで多数の動植物で解読されたゲノム配列をそれらの種の進化と照らし合わせることが可能となる。将来は、ヒトの起源を含め、生命進化の謎の解明に大きく貢献すると期待できる。
今後は、大気海洋研究所が進めるオーシャンDNAプロジェクトでORTHOSCOPE* を活用し、海洋生物・分布マップの発展を目指す。
発表雑誌
雑誌名:「Molecular Biology and Evolution」(2021年10月18日付)
論文タイトル:ORTHOSCOPE*: a phylogenetic pipeline to infer gene histories from genome-wide data
著者:Jun Inoue*
DOI番号:10.1093/molbev/msab301
アブストラクトURL:https://academic.oup.com/mbe/advance-article/doi/10.1093/molbev/msab301/6400256![]()
問い合わせ先
井上 潤
jinoue◎g.ecc.u-tokyo.ac.jp ※アドレスの「◎」は「@」に変換してください
用語解説
- 注1. ゲノム
- 生物が持つ1セットの遺伝情報の総和。その生物全てを構成する設計図。
- 注2. 遺伝子の水平伝搬
- 異なる種の遺伝子が侵入してゲノムに付加されたり、その一部が置き換わったりすること。通常遺伝子は、種ごとに世代から世代へと垂直に伝えられる。
- 注3. 全ゲノム重複
- 生物の進化の過程において、ゲノムを構成する染色体セットならびに保持される遺伝情報がコピーされて倍加すること。
- 注4. 環境DNA解析
- 水中に生息する生物の分布を、環境中に放出された DNA 配列を指標として把握する解析。
![]()